Malgré le développement des outils numériques, l'usage du papier pour écrire reste omniprésent dans le monde. La production annuelle mondiale de pâte à papier est de 178 Mt et celle de la France est de 1,6 Mt.
L’histoire du papier débute en Chine : Tsaï Loun ministre de l’Agriculture chinoise au IIIe siècle avant notre ère décrit la fabrication du papier à partir de l’écorce de bambou, de lin, de chanvre. Puis c’est le chimiste français Anselme Payen qui isole la cellulose sous forme de fibres blanches en 1838 [1].
Du bois aux pâtes à papier
Le bois est constitué essentiellement de trois polymères biosourcés étroitement imbriqués : la cellulose, les hémicelluloses et la lignine (fig. 1) dans des rapports 2/1/1 environ. La cellulose est un homopolymère linéaire constitué d’enchaînements de monomères de glucose associés par des liaisons éther (C-O-C) β-(1→4). Les hémicelluloses sont des hétéropolymères à chaînes constituées de différents monomères d'oses : glucose, xylose, mannose, arabinose, galactose, rhamnose et acides uroniques, ramifiées et entrelacées.
La lignine est un polymère tridimensionnel amorphe, réticulé de manière aléatoire constitué principalement d’unités phénoliques responsables de la couleur du bois et des pâtes à papier.

Figure 1. Principaux polymères biosourcés présents dans le bois. Crédit : Nathalie Marlin
Il existe deux sortes de pâtes à papier.
- 10 % sont d’origine mécanique et sont obtenues par meulage, une opération de défibrage mécanique. La pâte mécanique contient donc les trois polymères principaux du bois dans les mêmes proportions. Le rendement de production de la pâte mécanique est de 95%.
- 90 % sont d’origine chimique et sont issues du procédé kraft qui consiste en une cuisson de copeaux de bois en présence d’un mélange de soude et de sulfure de sodium réalisée environ à 150 °C pendant 4 à 6 heures. La lignine est dépolymérisée et passe en solution cette solution s’appelle la liqueur noire de par son aspect très foncé. Les fibres de cellulose ainsi libérées par voie chimique sont obtenues avec un rendement de 50%. Le restant de la matière (lignine et une partie des polysaccharides) passe dans la phase liquide : liqueur noire. Cette dernière est traitée de façon à régénérer les ions sulfure ainsi que la soude, puis sa combustion sert à produire l’énergie qui alimente l’usine. La pâte contient majoritairement de la cellulose et une partie des hémicelluloses mais une partie de la lignine, la lignine résiduelle, subsiste, et donne la couleur aux pâtes papetières non blanchies [2].
Blanchiment de la pâte à papier
Le blanchiment de la pâte mécanique consiste à décolorer les groupements chromophores de la lignine.
Pour cela on utilise des oxydants qui cassent certaines liaisons C=C présentes dans les cycles aromatiques de la lignine pour donner des liaisons C=O, ce qui entraine la disparition de la conjugaison des liaisons insaturées et donc de la couleur. La lignine décolorée reste dans la pâte après blanchiment, ceci explique le bon rendement de l’opération. Le principal agent de blanchiment de ces pâtes est l’eau oxygénée (H2O2). Après oxydation on obtient une pâte claire, la blancheur obtenue est suffisante pour produire du papier journal (60% norme ISO).
Le blanchiment de la pâte chimique consiste à retirer la lignine résiduelle de la pâte (4 à 8%) par dépolymérisation.
Le dioxygène et le dioxyde de chlore (ClO2) sont aujourd’hui les oxydants les plus utilisés avec l’eau oxygénée en complément.
L’oxygène est un agent de blanchiment sans impact pour l’environnement. Il est utilisé sous pression en milieu basique mais il est peu sélectif (uniquement 50% de lignine résiduelle est éliminée) de sorte que la cellulose est aussi en partie dégradée pendant l’opération.
Le dioxyde de chlore, classiquement utilisé en milieu acide, a une action sélective sur la lignine. Cette dernière est oxydée et dépolymérisée. Des produits organiques chlorés ayant une toxicité certaine sont produits en parallèle. C’est pourquoi un brevet récent (2018) propose d’utiliser le dioxyde de chlore en milieu alcalin pour réduire la production des produits polluants. Un protecteur de la cellulose doit être ajouté car en milieu basique le dioxyde de chlore est moins sélectif et dépolymérise aussi la cellulose [3].
Les pressions environnementales accélèrent les recherches et des procédés enzymatiques sont à l’étude.
On ajoute parfois, dans la suspension fibreuse de la pâte à papier, des charges minérales comme du dioxyde de titane pour augmenter la blancheur du papier et du carbonate de calcium pour obtenir une surface moins rugueuse afin d’améliorer l’impression.
Presque plus de 60% des papiers et cartons sont recyclés. Après collecte du papier et désencrage, on effectue les mêmes opérations de blanchiment si le papier recyclé exige un niveau de blancheur important.
Par ailleurs la liqueur noire obtenue dans le procédé kraft peut être mieux valorisée. Il existe de nombreuses voies de valorisation, par exemple elle peut être traitée par un procédé thermique en atmosphère inerte afin d’obtenir des matériaux carbonés graphitiques utilisés comme électrodes de supercondensateur ou capables de stocker de l’hydrogène [4].
L'auteur, Jean-Pierre Foulon, tient à remercier vivement Madame Nathalie Marlin, Maitresse de Conférence HDR de l’université Grenoble Alpes, pour son aide et sa relecture bienveillante à cette note pédagogique.
Pour en savoir plus
[1] La chimie et l’industrie papetière, O. Naef, IUPAC 2011 Chimia (2011) 65, 444, DOI: 10.2533/chimia.2011.444
[2] Le blanchiment de la pâte à papier a toujours la fibre de l’innovation... N. Marlin, J. Marcon, G. Mortha, A. Burnet, L’Actualité Chimique (mai-juin 2023) N° 484-485, 45
[3] N. Marlin : communication personnelle
[4] La liqueur noire, R. Backov, J.L. Bobet J. Olchowka, L’Actualité Chimique (mars 2024) N°493, 62
Crédit figure : Nathalie Marlin
Les abeilles sont indispensables par le service qu'elles rendent à la pollinisation : sans leur présence, nous perdrions 70 % des espèces cultivées en Europe. Mais leur disparition est bien évidemment préjudiciable à la production du miel. La France ne produit que le tiers de ses besoins en miel qui sont de 40.000 tonnes/an. Elle est donc obligée d'en importer. Or le miel est un produit onéreux : il est donc tentant pour les fournisseurs de proposer des produits de moindre coût de revient en les présentant comme des miels de qualité et d'origine conformes. C'est ce qu'on appelle l'adultération du produit. Il est important de contrôler la qualité des miels sur notre marché (1).
Le miel : un peu d'histoire
Son histoire (2) est très ancienne : le mot vient du latin mel, lui-même dérivé du grec μελί (meli). L'abeille est apparue sur Terre il y a environ 80 millions d'années, et le miel est le premier produit sucré découvert par l'espèce humaine. Les hommes préhistoriques le trouvaient dans les troncs d'arbres ou sous des rochers. Les civilisations les plus anciennes ont adopté le miel, lui attachant systématiquement une grande portée symbolique. Ainsi, les sumériens et les habitants de Babylone l’utilisaient au cours de leurs cérémonies religieuses, tout comme les Égyptiens qui s’en servaient pour l’embaumement des morts. L'abeille était alors considérée comme une véritable « messagère des dieux ». Dans certaines cultures, le miel était considéré comme l’aliment des aliments. Dans l'Odyssée, dès qu'Ulysse est accueilli quelque part, on lui apporte du miel, des olives et du fromage. Beaucoup voyaient en lui un véritable élixir de longue vie, et de nombreuses propriétés médicinales lui ont été prêtées au fil des âges. Il était également utilisé pour la conservation des aliments. Par exemple, au Ve siècle, l’historien Hérodote rapporte que les Grecs chassaient les faisans dans ce que l’on nomme aujourd’hui la Géorgie, et les immergeaient dans des amphores de miel pour le voyage de retour. On utilise aussi de la cire, par exemple pour imperméabiliser les récipients.
Ce n’est qu’à la fin du XVIIIe siècle qu’apparaît la ruche à cadres amovibles, qui permet à l’apiculteur de ne prélever qu’une partie des réserves sans anéantir la colonie d’abeilles. C’est une révolution. Au fil du temps, l’être humain a appris à sélectionner les miels en fonction des dates de floraison, pour obtenir des parfums très variés. Ainsi, on peut aujourd’hui répertorier plusieurs dizaines de miels de qualités organoleptiques différentes (trèfle, lavande, thym, sapin, montagne...).
Comment les abeilles fabriquent-elles le miel ?
Le miel (3)(4) est une substance sucrée élaborée par les abeilles à miel à partir du nectar (i). L'élaboration du miel commence dans le jabot de l'ouvrière, pendant son vol de retour vers la ruche. L'enzyme invertase (ii) présente dans le jabot, catalyse l'hydrolyse du saccharose qui donne du glucose et du fructose (fig. 1). Arrivée dans la ruche, la butineuse régurgite le nectar à une receveuse, qui le régurgitera et le ré-ingurgitera en le mêlant à de la salive et des sucs digestifs. Il est alors stocké dans les alvéoles et déshydraté par la ventilation assurée par certaines abeilles. Il arrive à maturité lorsque la concentration en eau est inférieure à 18 %, et sa durée de conservation est alors extrêmement longue. Pour produire 500 g de miel, les abeilles doivent effectuer plus de 17 300 voyages, visiter 8,7 millions de fleurs, rapporter 2 kg de nectar, le tout représentant plus de 7 200 heures de travail. Elles l'entreposent dans la ruche et s'en nourrissent tout au long de l'année, en particulier lors de périodes climatiques défavorables.

Quelle est la composition du miel ?
Le Codex alimentaire (5) donne les compositions maximales ou minimales en différents composés chimiques qui elles-mêmes dépendent de l’origine florale et géographique du miel. Ainsi par exemple pour un miel de nectar (iii), non pressé, et de composition florale « classique » (pas de lavande, ni de bruyère, ni de luzerne ou d’eucalyptus) le pourcentage d’eau ne doit pas dépasser 20 % ; tandis que la teneur de fructose et de glucose réunis doit être supérieure à 60 % et que celle en saccharose doit être inférieure à 5 % ; la teneur en matières insolubles dans l’eau doit être inférieure à 0,1 %.
Mais d’autres glucides (iv) se retrouvent dans le miel (on a en très souvent une teneur proche de 80 %) et leur présence ainsi que leur concentration permettent de déterminer l’origine florale ou géographique du miel.
Le miel contient aussi des protéines, des enzymes et des minéraux.
Les pollens permettent aussi de déterminer l’origine (6)(7).
Le miel ne contient pas de lipides et les quantités de résidus de pesticides, d’antibiotiques doivent être inférieures à des doses définies par la Commission européenne (8).
| Composés | Quantité (g/100 g) |
| eau | < 20 |
| Sucres Fructose + glucose saccharose | 79,8 > 60 < 5 |
| Matières insolubles | < 0,1 |
Quels sont les producteurs mondiaux du miel ?
Dans le monde, le premier producteur est la Chine, suivie de la Turquie, de l'Argentine, de l'Iran.
Le principal producteur de miel de l'UE est la Roumanie, suivie de l'Espagne, la Hongrie, l'Allemagne, l'Italie, la Grèce, la France et la Pologne. On trouve en France des miels AOP (Appellation d'origine protégée : Corse, sapin des Vosges) et IGP (Indication géographique protégée : Alsace, Provence).
Les fraudes suspectées sur le miel
Environ 46 % des miels importés dans l'Union européenne, principalement en provenance de Chine et de Turquie, seraient frauduleux, en particulier dilués pour en diminuer le prix et en augmenter la quantité et beaucoup contiendraient des produits toxiques (pesticides, en particulier néonicotinoïdes, chloramphénicol, métaux lourds).
D'autre part, les produits importés, en particulier de Chine et de Turquie, sont réexportés comme produits locaux, si bien qu'un tiers des miels dans l'Union européenne ne seraient pas conformes à la provenance indiquée.
En résumé les fraudes peuvent présenter plusieurs formes :
- la présence d'impuretés (des résidus de pesticides par exemple) ;
- l'existence d'une dilution avec des sirops de sucre ;
- l'ajout d'additifs non déclarés (du sucre brut, du sucre raffiné, du sirop de maïs à forte teneur en fructose (HFCS) (v), du sucre de palme ;
- avoir une fausse origine botanique (miel « de lavande » par addition d'essence de lavande à un miel ordinaire) ;
- avoir une fausse origine géographique et contenir des miels bon marché (de Chine et autres) ;
- un étiquetage insuffisant. En effet, l'étiquetage (vi) est important et est rarement respecté.
Quelles méthodes employer pour contrôler les miels ? (9)
Il faut donc vérifier si ces miels sont :
- conformes à la réglementation
- respectueux de l'environnement
- authentiques
- sans danger
Il y a longtemps que le contrôle de la qualité et l'authenticité des aliments est pratiquée. L’identification et le dosage des éléments présents se font principalement par des méthodes chromatographiques.
Recherche de résidus (10)
Ainsi par exemple la chromatographie liquide couplée en tandem à la spectrométrie de masse (LC-MS/MS) et la chromatographie gaz couplé à un temps de vol pour l’analyse en spectrométrie de masse (GC-ToF) permet de déterminer une grande quantité de résidus de pesticides (insecticides, acaricide…) ou d’antibiotiques. Ainsi on peut détecter des résidus de coumaphos, utilisé auparavant (mais maintenant interdit en France) pour lutter contre le Varroa, parasite bien connu des apiculteurs. En France, c’est le Service Commun des Laboratoires et plus particulièrement celui de Marseille qui recherche ce type de composés dans le miel.
La fraude à la dilution par ajout de sirop de sucre (11)
Suivant le type de plante, le rapport isotopique 13C/12C diffère. Ainsi le miel provenant de plantes florales aura un δ13C réf de l’ordre de -28 ‰ alors que celui du sirop de maïs ou de canne à sucre est de l’ordre de -14 ‰. Ainsi une analyse élémentaire par spectroscopie de masse de rapport isotopique (EA-IRMS) peut indiquer une addition d’un sirop de sucre au miel. Pour affiner la mesure on détermine aussi la différence des rapports isotopiques des glucides (majoritaires dans le miel) et des protéines qui seront d’autant plus différents que la quantité de sirop ajouté sera grande.
La fraude à l’origine botanique ou géographique
Cela consiste à vendre un miel soi-disant issu de telle fleur et de tel lieu, mais qui est en réalité mélangé à un miel (moins cher) d’une autre provenance. Pour la mise en évidence de ce type de fraude, on utilise des spectres de résonance magnétique du proton (RMN 1H) (12), ou proche infrarouge (NIR), ou des chromatogrammes par détection UV-Visible après une chromatographie liquide haute performance ((HPLC-UV) (13). On utilise alors des méthodes non ciblées qui analysent l'ensemble du produit sans a priori, et qui permettent de détecter des fraudes non connues. On compare les résultats à des bases de données produites à partir d'échantillons connus et traçables. Le traitement de toutes ces données est facilité par l'apport de l'Intelligence Artificielle. Les méthodes utilisées sont des méthodes non supervisées telles que l’analyse en composante principale (PCA) ou les analyses discriminantes par les moindres carrés partiels (PLS-DA) (vii) (13). On pourra ainsi détecter l'ajout de substances de charge (sirop de sucre), d'altérations (ajout d'autres miels), et même le contrôle de l'origine géographique.
Enfin pour déterminer la provenance d’un miel, on peut aussi étudier les pollens contenus dans le miel et comparer ces pollens à la composition florale du miel. Ceci est ce qu’on appelle la mélissopalynologie (viii) (7).
L’identification des sucres présents
D’autres méthodes ciblées complètent le tableau ; ainsi la chromatographie à échange d’anions à détection ampérométrique (HPAEC-PAD) (6) ou des mesures enzymatiques ou des mesures de photométrie permettent de déterminer les quantités de chacun des sucres présents (glucose, fructose, maltose, HMF (ix) …).
Les laboratoires
Il existe des laboratoires spécialisés dans ces contrôles, comme le Centre de Compétence Authenticité Eurofins de Nantes (14)(15). Sur dix produits susceptibles d'adultération, le miel est en bonne place, avec l'huile d'olive, le vin, les épices, les cafés et thés. Historiquement, c'est en 1981 qu'a été inventée à Nantes la méthode SNIF-NMR® (Site-specific Natural Isotopic Fractionation studied by deuterium Nuclear Magnetic Resonance) pour le vin et l'alcool. Le laboratoire Eurofins a été créé en 1987, FINS étant l'acronyme français de SNIF.
L’ANSES (Agence nationale de sécurité sanitaire de l’alimentation, de l’environnement et du travail) a un dossier complet sur les abeilles (16).
C’est la combinaison de l’ensemble des analyses qui permet de déterminer la qualité d’un miel.
À propos de la disparition des abeilles
Depuis quelque temps, vous n'ouvrez plus votre poste de radio sans entendre parler du frelon asiatique : il serait de plus en plus envahissant et, par suite de son agressivité, très dangereux pour les abeilles.
Certains pesticides aussi sont responsables de la disparition des abeilles. Le cas des néonicotinoïdes est particulièrement préoccupant, car ils peuvent induire chez l'abeille des troubles de l'apprentissage et de la mémoire qui dégradent ou détruisent sa capacité à retrouver des aliments ou sa ruche, au point de parfois menacer la santé de la ruche entière. Ils sont suspectés de jouer un rôle clé dans l'effondrement mondial des populations de pollinisateurs domestiques et sauvages (17).
Et leur disparition est bien évidemment préjudiciable à la production du miel.

Photographie Bernd Amann (20/06/2015)
(i) Le nectar (du latin nectarium, breuvage des dieux !) est un suc secrété généralement par les nectaires, au cœur des plantes. Il constitue la matière première du miel. Les plantes produisant du nectar en abondance sont dites nectarifères et les plantes présentant un intérêt pour la production de miel sont dites mellifères.
(ii) L'enzyme inverse le pouvoir rotatoire [α] D20, qui passe de +66,5° pour le saccharose à -39,3° pour le mélange glucose (+52,7°) + fructose (-92°).
(iii) Il existe des miels de nectar mais aussi de miellat (miel de sapin, par exemple : le miellat est fabriqué par les pucerons sur les sapins).
(iv) Les glucides (ou saccharides) sont composés de monosaccharides (glucose, fructose, galactose…) de disaccharides (maltose, saccharose…), trisaccharides (mélézitose, raffinose…), polysaccharides.
(v) HFCS = High Fructose Corn Sirup
(vi) Détail des réglementations sur l’étiquetage : • Origine, provenance • Dénomination de vente complète • Origine florale • Origine territoriale • Critères spécifiques de qualité [Mode de production, AOP, IGP; et d’autres termes (crémeux, de printemps…)] • Quantité nette• Date de Durabilité Minimale et/ou N° de lot • Nom ou raison sociale et adresse de l’exploitant.
(vii) PCA : analyse en composante principale et PLS : Partial Least Squares ; ces méthodes consistent à réduire les dimensions des données ; par exemple à partir d’un spectre d’1 miel, on obtient un couple de point (X,Y). On trace ensuite pour l’ensemble des N miels donnés, les N points (X,Y) et on cherche si des clusters se forment et s’ils correspondent à une propriété donnée (par exemple miel de sapin ou miel de Corse…).
(viii) du grec mel, miel et palunein, répandre de la farine (ici le pollen ou les spores).
(ix) HMF : Hydroxy-Méthyl-Furfural produit de la déshydratation du fructose
Références : merci à J-P Dal Pont pour de précieux numéros des annales de la Société des Experts Chimiques de France
(1) Marcincal A., Royer M., (2022) Le miel, un produit naturel. L'abeille, un enjeu stratégique ?, Annal. Fals. Exp. Chim. Tech. N°996, p74.
(2) L’histoire du miel (site Les rochers du Tigou) https://www.tigoo-miel.com/le-miel-et-son-histoire/
(3) Miel sur Wikipedia
(4) Boutonnier, J.-L., (2023) Miels, Techniques de l'ingénieur (Janvier 2023)
(5) Norme pour le miel (PDF) – source OMS 2022, NORME POUR LE MIEL CXS 12-1981 dernière modification 2022
(6) Michel, Killian, Détection des fraudes en sucres dans les miels : appui au développement d'une méthode se basant sur la technique HPAEC-PAD. Ecole polytechnique de Louvain, Université catholique de Louvain, 2023. Pages 19 et 20 Prom. : Luis Alconero, Patricia ; Massaux, Carine. https://hdl.handle.net/2078.1/thesis:42099
(7) Lobreau-Callen D., Clement M-D., Marmion V., Les miels, Techniques de l'ingénieur Juin 2000
(8) Journal Officiel UE Limites maximales de résidus de pesticides 2017 (PDF)
(9) Guyon F., Landuré M., Gaudefroy M., Angioni C., Fino L., Le contrôle et le développement de nouveaux outils des contrôles des miels : un enjeu stratégique salutaire pour la filière française ! SECF webinaire MIEL Episode 1, 4 avril 2022
(10) Wiest, L., & Vulliet, E., Analyse multi-résidus de pesticides dans le miel : enjeux et défis analytiques. In Annales des falsifications, de l'expertise chimique et toxicologique, Vol. 985 (2016) pp. 17-27
(11) Official Methods of Analysis (OMA) par International Association of Official Agricultural Chemists, AOAC 9941-41 et AOAC 978-17
(12) Rhee Y., Shilliday E.R., Matviychuk Y., Nguyen T., Robinson N., Holland D.J., Connolly P.R.J. and Johns M.L., Detection of honey adulteration using benchtop 1H NMR spectroscopy, Anal. Methods 15 (2023) 1690 https://doi.org/10.1039/D2AY01757A
(13) Egido C., Saurina J., Sentellas S., Núnez O., Honey fraud detection based on sugar syrup adulterations by HPLC-UV fingerprinting and chemometrics, Food Chemistry 436 (2024) 137758 https://doi.org/10.1016/j.foodchem.2023.137758
(14) Thomas F., (2022) Analyse non ciblée en authenticité : l'IA pour la gestion des bases de données et l'interprétation dans le contrôle alimentaire, Annal. Fals. Exp. Chim. Tech. N°996, p.48
(15) Thomas F., (2022) Comment lutter contre la fraude économique et préserver l'authenticité des miels ? SECF webinaire MIEL Episode 1, 4 avril 2022 https://www.la-sca.net/actualites-apicoles-142/sorties-conferences-expositions/conference-le-miel-un-produit-naturel-l-abeille-un-enjeu-strategique
(16) Les abeilles, des pollinisateurs essentiels dont la santé est menacée - ANSES anses.sante-des-abeilles
(17) Mitchell E.A.D, Mulhauser B., Mulot M., Mutabazi A., Glauser G. & Aebi A., A worldwide survey of neonicotinoids in honey, Science, Vol. 358 (2017) issue 6359, pp. 109-111 https://doi.org/10.1126/science.aan3684
Crédit illustration : Bernd Amann (20/06/2015).
Pourquoi des cristaux de synthèse ? Longtemps les cristaux naturels trouvés dans la roche, pierres rares, transparentes, incolores ou colorées par des impuretés, ont été utilisés principalement en joaillerie.
Sous forme de monocristal, un composé peut posséder des propriétés physico-chimiques particulières et depuis longtemps les chercheurs ont recours au monocristal pour accéder à la structure et aux propriétés qui en découlent. Certains cristaux sont désormais indispensables dans des applications modernes : cristaux de quartz (piézoélectricité), cristaux de silicium ou de germanium (semi-conducteurs), cristaux utilisés pour les besoins de l’optique (transparence, biréfringence), de l’électronique, des lasers solides… L'électronique et l'informatique s’appuient sur les propriétés de cristaux ultra-purs.
Or les cristaux naturels (pierres précieuses, quartz, etc.) ont souvent des défauts et/ou sont issus de processus géologiques de plusieurs millions d’années. Leur rareté mais aussi l’impact environnemental et économique de leur extraction les rendent très coûteux.
De nombreuses techniques de synthèse ont été développées pour obtenir, dans des conditions de croissance reproductibles, des cristaux ultra-purs (ou volontairement dopés par des atomes spécifiques), exempts de défauts et répondant aux exigences industrielles (dimension, densité de défauts, homogénéité, orientation, couches minces, etc).
Cristallisation
La cristallisation est le processus de formation d'un cristal. C’est le passage des atomes d'un état plus ou moins désordonné (liquide, gazeux, solide vitreux ou amorphe) à un état ordonné à plus ou moins longue distance. Cela requiert du temps et de l’énergie, pour faire migrer les atomes vers leur position finale, dans le réseau ordonné.
Plusieurs méthodes existent pour obtenir un monocristal de synthèse (cristallogenèse), selon la température de fusion, la fusion congruente ou non, le changement de structure en fonction de la température etc. Les principales méthodes sont basées sur :
- le refroidissement lent d’un composé en fusion : méthodes de Verneuil (1), Czochralski (2), zone fondue verticale ou four à image (3);
- le dépôt à partir d’une solution sursaturée d’un composé : croissance hydrothermale (4) à partir d’une solution aqueuse, méthode des flux à partir de sels fondus (5);
- le dépôt en phase vapeur.
 |  |
| Figure 1 : Procédé Verneuil. Schéma Benoît Grosjean.Site CultureSciences Chimie licence CC-BY-NC-SA | Figure 2 : Procédé Czochralski. Damien Sangla. Nouveaux concepts pour des lasers de puissance : fibres cristallines dopées Ytterbium et pompage direct de cristaux dopés Néodyme, Thèse Université Claude Bernard Lyon 1 (2009) ⟨tel-00448320⟩ |
 |  |
| Figure 3a : Four à image. Equipe Matériaux pour la Photonique et l'Opto-électronique, Chimie ParisTech, CNRS, Institut de Recherche de Chimie Paris | Figure 3b : Four à image. Equipe SP2M- ICMMO, Université Paris-Saclay |
Quelques cristaux de synthèse à vocation technologique
Les exemples de cristaux de synthèse largement utilisés permettent d’illustrer ces méthodes, développées d’abord en laboratoire puis à l’échelle industrielle.
Rubis (Tfus = 2050 °C)
Le rubis naturel est une pierre précieuse rouge, très rare, très dure et transparente. C’est de l’alumine contenant du chrome qui lui donne sa couleur rouge, Al2O3Cr.
- En raison de leur grande résistance à l'usure, les rubis sont depuis longtemps utilisés en horlogerie où ils améliorent l’efficacité des rouages mécaniques.
- En 1960 le rubis est la source du premier effet laser, un des seuls lasers émettant dans le visible, qui a depuis connu un développement ininterrompu, dans l’industrie, la médecine, la vie quotidienne, comme dans la recherche.
Pour ces raisons, les scientifiques ont très tôt cherché à obtenir le rubis en laboratoire. Les premières synthèses sont dues au chimiste français Verneuil, « inventeur » de la cristallogenèse (1891-1902). Souvent appelée Procédé Verneuil (1), sa méthode de fabrication est la croissance par fusion à la flamme, toujours largement employée aujourd'hui. Pour l’optique (lasers), on emploie des rubis synthétiques de très haute qualité, fabriqués par tirage vertical à partir d'un bain fondu, méthode dite de Czochralski (2). Le four à image ou four à concentration de rayonnement (3) est aussi utilisé pour produire des barreaux de rubis.
Diamant (Tfus = 3547°C à l’abri de l’air…)
Le diamant est une des variétés cristallisées du carbone dont la forme stable est le graphite. Il est constitué de carbone pur, on ne peut donc pas parler de « synthèse ».
Outre son fort pouvoir réflecteur, dû à son indice de réfraction élevé (2,42) qui lui donne un éclat unique, c’est le plus dur des minéraux connus, très utilisé en milieu industriel ; c’est un isolant électrique, ses propriétés mécaniques, optiques, thermiques et électroniques sont exceptionnelles.
- En 1772 Lavoisier démontre que le diamant est constitué de carbone, les premiers essais de synthèse commencent à la fin du 19e siècle (1880 - J. B. Hannay, 1893 - H. Moissan). La première véritable synthèse a lieu en 1954 (6).
- Les diamants de culture sont obtenus par technologie HPHT (Haute Pression, Haute Température) qui reproduit la formation des diamants naturels (7).
Le diamant peut aussi être produit par dépôt chimique en phase vapeur qui produit plus vite des diamants de meilleure qualité (8).
Quartz (Tfus = 1750°C)
Le quartz est une des variétés cristallines de la silice SiO2. Les cristaux de quartz sont piézoélectriques et largement utilisés dans l’horlogerie (montres à quartz), les sonars et pour définir les fréquences de fonctionnement des appareils électroniques (téléphones, ordinateurs, GPS…).
- Le quartz naturel (ou cristal de roche) est très abondant, mais présente de nombreux « défauts » tels que macles, dislocations, inclusions, changement de structure en fonction de la température etc. Il est donc nécessaire de le synthétiser.
- La cristallogenèse se fait par procédé hydrothermal, reproduisant les conditions naturelles de formation des cristaux de roche (4). C'est un processus lent qui peut durer plusieurs semaines.
Silicium (Tfus =1414°C)
Le silicium Si est encore le matériau essentiel de l'ère du numérique, même si d’autres éléments prennent la relève. Il est semi-conducteur et c’est un des constituants de base des panneaux solaires, transistors, circuits intégrés, microprocesseurs et autres dispositifs électroniques. La pureté requise varie selon l’application visée : pour les cellules solaires photovoltaïques, il faut du silicium de pureté 99,999 9%, alors que pour les puces électroniques il faut du silicium de pureté électronique, soit 99,999 999 99%.
- La matière première à partir de laquelle il est obtenu est la silice SiO2. Comme c’est une source abondante (environ 60 % de la croûte terrestre), l'industrie des semi-conducteurs a largement développé la synthèse de cristaux de silicium purs ou dopés à partir de la silice.
Dans un premier temps, la silice est réduite par du carbone (selon la réaction SiO2 + C → Si + CO2) puis le silicium polycristallin obtenu est purifié et recristallisé par fusion/ refroidissement (tirage Czochralski, ou zone fondue).
En particulier, le procédé par zone fondue permet de purifier des composés cristallisés stables à la fusion et d'obtenir de très hauts degrés de pureté (99,999 % en masse dans le cas du silicium). En effet, lors de la recristallisation, les impuretés restent préférentiellement dans la zone fondue.
Peut-on différencier les cristaux naturels de ceux obtenus en laboratoire ?
Le cristal synthétique présente, par définition, mêmes composition, structure et aspect que son équivalent naturel et les mêmes caractéristiques physiques et chimiques.
Mais chaque méthode laisse des marques de croissance, voire des impuretés (azote, carbone…) inconnues dans le matériau naturel, et des signes distinctifs qui peuvent être décelés à l'aide d'instruments optiques professionnels. D’autre part, les dimensions diffèrent, parfois fortement ! Alors que le plus gros diamant de laboratoire pèse 155 carats (9), le plus gros diamant naturel de joaillerie connu, le Cullinan, pesait 3 106 carats, soit 621 grammes ! Inversement, le rubis de synthèse peut atteindre plusieurs kilos et des dimensions de 20 à 30 cm.
Longtemps réticents envers les « gemmes » de synthèse, les grands joailliers commencent à les utiliser ! Toutefois on ne peut passer sous silence les réactions « contre » le diamant de synthèse, dont la fabrication est très énergivore et prive de ressources et d’emplois les pays riches en diamants naturels - ce dernier argument paraît contestable lorsqu’on connaît l’existence des diamants de sang.
Conclusion
L’évolution des domaines de haute technologie dépend clairement des monocristaux de synthèse. Les chimistes ont su « reproduire » la nature et les techniques n'ont cessé d’évoluer. Celles présentées ici ont de nombreuses variantes, qui dépendent du matériau et de l’application visée. Mais la recherche y recourt également pour étudier les relations entre structure et propriétés d’un composé (et les éventuelles applications).
Andrée Harari
(1) Procédé Verneuil, C. R. Acad. Sc. (1902) p. 791. Procédé Verneuil : le matériau en poudre fond dans une flamme de chalumeau oxhydrique avant de recristalliser lentement au contact d’un monocristal préalablement orienté qui sert de germe.
(2) Méthode Czochralski (1918) : J. Czochralski, Z. Physik Chem. 92 (1918) p. 219
Le matériau (par exemple Al2O3Cr) est fondu dans un creuset. Le liquide se solidifie sur un germe monocristallin de petite taille, suspendu au contact du liquide. On tire ce germe vers le haut tout en le faisant tourner lentement. La taille des cristaux obtenus peut atteindre plusieurs centimètres.
(3) Four à image (zone fondue verticale). Le rayonnement d’une source d’énergie (lampe, laser, soleil) est concentré, grâce à des miroirs, pour chauffer et fondre les matériaux passant dans ce faisceau. La migration lente et contrôlée hors de la zone chaude entraîne la recristallisation par refroidissement lent.
(4) Croissance hydrothermale. Dans un cylindre rempli d'eau, on dispose, par exemple, un cristal de quartz naturel (germe sur lequel le cristal artificiel va croître) et de la silice sous une forme soluble. L'ensemble est soumis à une forte pression (environ 100 MPa) et porté à 400 °C mais de sorte que la partie supérieure soit légèrement moins chaude. La solution saturée en silice qui se forme en partie basse est entraînée par convection vers le haut du récipient, où elle devient sursaturée (la solubilité décroît en fonction de la température). La silice précipite alors sous forme de quartz au contact du germe.
(5) W. G. Pfann, Principles of Zone Melting, Transactions of the American Institute of Mining and Metallurgical Engineers 194 (1952) p. 747.
(6) H.T. Hall, General Electric - U.S. Patent 2,947,608 or "Diamond Synthesis", Aug. 2, 1960
(7) Les diamants se sont formés il y a 2,5 milliards d’années, à 200 km sous la surface de la Terre, dans des conditions extrêmes de pression (5 GPa, environ 50 000 bars et de température (1500 °C environ). C’est leur expulsion brutale au sein du magma, lors des éruptions volcaniques, qui les a apportés à la surface.
Dans la méthode HTHP, un petit germe de diamant est placé dans une presse remplie de graphite et soumise à des températures de 1500 à 2000 °C et des pressions supérieures à 70 000 bar. Les atomes de carbone du graphite s’agrègent autour du grain de diamant et lui permettent de croître, très lentement. On obtient ainsi des cristaux de quelques carats (1 ct = 0,2 g).
(8) Dépôt chimique en phase vapeur. La technique utilise un mélange méthane + hydrogène, chauffé pour créer un plasma de carbone au-dessus d'un substrat, sur lequel les atomes de carbone se déposent pour former les couches cristallines successives.
(9) Schreck, M. et al., Ion bombardment induced buried lateral growth: the key mechanism for the synthesis of single crystal diamond wafers, Sci. Rep. 7 (2017) 44462; doi: 10.1038/srep44462
Pour en savoir plus
- B. Grosjean, Les procédés de cristallogenèse, piliers méconnus de la technologie moderne (2017) Site Culture Sciences Chimie ENS
- Procédés de fabrication des gemmes synthétiques, Site Gemmo.eu
- Méthodes d'élaboration de cristaux massifs pour l'optique, B. Ferrand, LETI/DOPT/SCOPI/LCDO/Matériaux/CEA Grenoble Collection SFO 8 (2003) 3-21
- J.-C. Bouilliard, Et l’homme créa la pierre. Les synthèses de cristaux. AMIS. 1996. voir https://www.amis-mineraux.fr/publications/toutes-les-publications.html
Crédit illustration : Un diamant synthétique taillé, conçu avec la méthode CVD. Steve Jurvetson - https://www.flickr.com/photos/jurvetson/156830367/, licence CC BY 2.0, Lien
Les radicaux sont des espèces chimiques possédant au moins un électron non apparié. La spectroscopie paramagnétique électronique (RPE) permet de détecter ces espèces radicalaires.
Structure des radicaux carbonés [1]
Il existe essentiellement quatre types d’espèces carbonées dans lesquelles l’atome de carbone n’est pas tétravalent : il s’agit des carbanions, des carbocations, des carbènes (R2C portant à la fois une lacune électronique et un doublet non liant) et des radicaux carbonés. Parmi eux seuls les carbanions respectent la règle de l’octet.
Exemples

L’activité optique d’une molécule ne possédant qu’un atome de carbone asymétrique est « perdue » lorsqu’on forme le radical carboné associé : on peut considérer alors que la structure devient plane. Cependant des mesures par RPE montrent que le radical tert-butyle est pyramidal avec une barrière d’inversion très faible, autour de 7-8 kJ.mol-1. On peut donc dire qu’à température ambiante le radical carboné apparaît « dynamiquement » plan, autrement dit tout se passe comme s’il était plan !
Il en est de même pour les radicaux éthyléniques Z ou E qui sont en équilibre et ne conservent pas l’information de la configuration Z ou E.

Figure 1
Stabilité des radicaux carbonés [2]
- Les radicaux sont des espèces très réactives avec en général une durée de vie très courte. Par exemple le radical méthyle a une demi-vie de l’ordre de 10 minutes. Cependant quelques radicaux sont stables, citons le radical (C6Cl5)3C▪ qui est stable pendant plusieurs jours en solution et stable sous forme solide.
- Un radical peut être stabilisé par la présence d’hétéroatomes adjacents possédant un « doublet non liant » : l’interprétation est la stabilisation par la délocalisation électronique dans le cadre de la théorie des orbitales moléculaires entre l’orbitale à deux électrons de l’hétéroatome et l’orbitale moléculaire occupée par 1 électron de l’atome radical.

Figure 2
Source des radicaux [3]
Si la rupture homolytique d’une liaison donne naissance a priori à deux espèces moléculaires radicalaires, les conditions expérimentales sont difficiles à mettre en œuvre. On peut employer un amorceur comportant une liaison fragile thermiquement permettant d’initier ultérieurement la rupture de la liaison souhaitée. On peut aussi provoquer la rupture par apport d’énergie sous forme de lumière à des longueurs d’onde bien spécifiques comme le montrent les exemples suivants :
- les composés peroxydes possédant une liaison O-O de faible enthalpie de liaison de l’ordre de 130 kJ.mol-1 conduisent à des radicaux par la cassure de cette liaison vers 100°C ;
- les composés azoïques possédant une liaison N=N se rompent dès 60°C ;
- les molécules avec une liaison Sn-Sn sont décomposées à l’issue d’une excitation lumineuse ;
- les composés stanneux avec une liaison Sn-H, comme l’hydrure de tributylétain Bu3SnH, découvert par D.Barton (Prix Nobel de chimie en 1969) permettent l’obtention d’un radical Bu3Sn▪. Ce composé a été très employé, mais sa toxicité observée lors d’expositions prolongées a entraîné une diminution progressive de son utilisation ;
- on a utilisé depuis des composés contenant du bore en particulier des alkylboranes, tels que Et3B, qui en présence du dioxygène de l’air, conduisent à la formation du radical éthyle (Et▪) ;
- ou encore des composés contenant du silicium tels que H-SiCl3 mais nécessitant l’emploi d’un catalyseur de photodissociation à l’iridium ;
- actuellement des décharges dans des plasmas (par onde sonore, laser ou UV) permettent de générer des espèces radicalaires à pression atmosphérique et à température ambiante sans amorceur !
Principe de la réactivité des radicaux [4]
De nombreuses réactions radicalaires font intervenir un processus mécanistique « en chaîne » qui est un mécanisme à séquence fermée comportant trois étapes principales : l’amorçage, la propagation et la terminaison (par recombinaison). Il est important de mentionner aussi les réactions de transfert d’un radical à une autre espèce chimique moléculaire présente dans le milieu réactionnel.
Il est intéressant de prévoir approximativement la réactivité des radicaux par la théorie des orbitales frontalières : l’électron célibataire occupe l’orbitale moléculaire la plus haute appelée SOMO (en anglais Singly Occupied Molecular Orbital) selon les règles de remplissage de Pauli. L’expérience montre qu’il existe des radicaux nucléophiles et des radicaux électrophiles.
Dans le cadre de la théorie des orbitales frontalières un radical possédant un groupe électrodonneur (ou respectivement électroattracteur) a une SOMO rehaussée (ou respectivement abaissée) et possède alors un caractère nucléophile (ou électrophile). Un radical nucléophile (ou électrophile) réagira de préférence avec une molécule par l’intermédiaire de sa BV (ou sa HO) d’une autre molécule, et ceci d’autant plus si les deux niveaux d’énergie des deux orbitales sont proches : c’est en particulier ce qui explique la réactivité des radicaux avec les composés éthyléniques activés par des groupes attracteur ou donneur. Par exemple dans le styrène le groupe phényle est électrodonneur.

Figure 3
Cependant il existe des composés éthyléniques qui possèdent, à la fois sur un des deux atomes de carbone de la double liaison, un groupe attracteur et un autre donneur d’électron qui sous l’action d’un amorceur conduisent à des radicaux stabilisés par délocalisation électronique par exemple

Figure 4
On parle alors d’effet captodatif selon la nomenclature de Viehe qui a isolé de tels radicaux en 1988 à l’Université de Louvain en Belgique.
Un point anecdotique mais révélateur de l’intérêt des radicaux : la réaction du chlorure de méthylmagnésium dans le solvant THF sur de l’éthanal, traditionnellement expliquée par une coupure hétérolytique C-Mg, est plutôt envisagée actuellement par un mécanisme à plusieurs étapes avec en particulier une coupure homolytique de la liaison carbone-métal sur un état intermédiaire parmi les états intermédiaires et de transition, provenant de travaux de simulation théorique. Cette interprétation provient notamment des travaux de O. Eisenstein à Montpellier publiés en 2020 soit plus de cent vingt ans après la découverte de cette réaction par Victor Grignard !
Polymérisation radicalaire [4]
La polymérisation des alcènes est difficile à contrôler à cause des réactions de transfert d’un radical au polymère et/ou au monomère qui créent des ramifications lors de l’allongement de la chaîne. Cependant depuis vingt ans, des travaux ont permis de réaliser une polymérisation radicalaire contrôlée par transfert de chaîne réversible par addition-fragmentation appelée Reversible Addition- Fragmentation Chain Transfer (RAFT) en anglais. Les principaux agents de transfert sont des dérivés soufrés du type

Figure 5
Ainsi S. Zard (École Polytechnique) en France a développé en collaboration avec Rhodia, devenu depuis Solvay, la technologie MADIX (en anglais Macromolecular Design by Interchange of Xanthates) qui met en jeu des xanthates S (C=S) OR. Ces derniers permettent alors la formation du radical R▪ qui a une durée de vie suffisante lui permettant de réagir même avec un alcène non activé par une fonction. Les résultats excellents obtenus au laboratoire ont entraîné le dépôt de plusieurs centaines de brevets depuis 1997 ! Les applications sont multiples dans le domaine des adhésifs, des traitements de surface, tensioactifs et cosmétiques. Par exemple l’équipe de S. Zard a mis au point et la société Rhodia a commercialisé des copolymères diblocs employés principalement dans le domaine du traitement de l’eau.
Formation des molécules dans le milieu interstellaire (MIS) [5], [6]
Le MIS englobe tout l’espace qui se trouve entre les étoiles ou à leur voisinage.
Une partie des molécules interstellaires observées dans le MIS résultent de chaînes de réactions impliquant des radicaux en phase gazeuse. Mais certaines molécules nécessitent la présence de grains de poussière interstellaires pour activer des réactions chimiques entre les atomes à leur surface (catalyse hétérogène). L’omniprésence de la molécule de dihydrogène, avec une densité allant de 100 à 108 atomes d’hydrogène par cm3, s’explique par la dissociation du dihydrogène en atomes sous l’excitation du fort rayonnement solaire suivie par une recombinaison en molécules sur les grains de poussière dans des zones non illuminées par le rayonnement solaire.
Le MIS contient de nombreuses molécules possédant l’élément carbone. Pour des photons d’énergie suffisante, l’étape photochimique de départ engendre des radicaux qui peuvent entrer dans des chaînes de réaction conduisant à des molécules de plus en plus complexes jusqu’à des systèmes dits prébiotiques tels que les acides nucléiques comme la cytosine et l’uracyle.
Signalons que le di-radical éthynyle de formule  noté C2▪▪ a été observé dès la fin du XIXe siècle dans les comètes ; en particulier Jackson en 1976 a montré dans la comète de Halley l’existence de l’acétylène (éthyne) qui provient du radical C2▪▪, observé en spectroscopie en lumière visible lors de phénomènes de relaxation.
noté C2▪▪ a été observé dès la fin du XIXe siècle dans les comètes ; en particulier Jackson en 1976 a montré dans la comète de Halley l’existence de l’acétylène (éthyne) qui provient du radical C2▪▪, observé en spectroscopie en lumière visible lors de phénomènes de relaxation.
Radicaux non carbonés
Rappelons que le dioxygène moléculaire est un diradical moléculaire dans son état stable ! Le remplissage des orbitales moléculaires selon les règles de Pauli et de Hund conduit en effet à l’occupation de chacune des deux orbitales antiliantes de type pi. Ainsi l’oxydation de l’isopropylbenzène par l’oxygène de l’air donne le radical cumyle (Me)2 C▪-Ph qui conduit au phénol et à l’acétone, procédé réalisé industriellement.
L’exemple le plus important est la molécule NO. C’est une espèce radicalaire stable possédant une double liaison entre N et O et un électron non apparié. Il a été mis en évidence dès 1986 dans plusieurs systèmes biologiques par des mesures de spectrométrie de masse et de chimiluminescence. Dans la nature il peut réagir avec le dioxygène moléculaire (diradical !) ou des métaux de transition engagés dans des molécules complexes telles que l’hémoglobine. La durée de vie de quelques secondes des radicaux intermédiaires conduit à des réactions très rapides rendant difficile la détection dans les milieux biologiques. Le monoxyde d’azote NO a une action antibactérienne (également sur des cellules tumorales) et antiparasitaire. Cependant, il ne faut pas oublier que NO est un polluant présent dans la fumée de cigarette ou dans certains milieux urbains et industriels. En effet, à des concentrations de 100 ppm dans l’air il altère les tissus pulmonaires ; sa toxicité provient de son oxydation par la molécule de dioxygène qui conduit au dioxyde d’azote NO2 réputé très toxique !
Note : Je tiens ici à remercier vivement Julien Lalande pour ses nombreuses corrections de syntaxe ainsi que les formules des molécules et aussi pour les belles discussions sur le sujet !
Bibliographie :
[1] Cours de J.Y. Lallemand de l’École polytechnique : communication personnelle (1998)
[2] Livre de J. Fossey et coll. : les réactions radicalaires en chimie organique (1995) chez Masson
[3] L. Fensterbank – Nouvelles catalyses pour accéder à la complexité moléculaire article téléchargeable de l’Actualité Chimique, N° 435 décembre 2018, pages 13 et suivantes
[4] S. Zard et coll., Une quête de nouvelles réactions pour la synthèse organique article téléchargeable de l’Actualité Chimique, N° double 393-394 février-mars 2015 page 48 et suivantes
[5] Coordinateurs S.Leach et E. Amouyal, Molécules et interstellaires et photochimie dans l’espace, dossier téléchargeable de 4 articles de l’Actualité Chimique N° 315 janvier 2008 pages I à XXIII
[6] Michel Guelin, Molécules dans l’Univers : Où ? Quand ? Comment ? Pourquoi ? conférence et article, site Mediachimie.org, Colloque Chimie, aéronautique et espace du 8 novembre 2017, Chimie, aéronautique et espace (2018) EDP Sciences, ISBN9782759822836 p. 183
Crédit illustration : image par PublicDomainPictures de Pixabay
La bactérie
Une bactérie [1] est une cellule très simple, « un sac » qui renferme tout ce qui est nécessaire à sa survie (fig. 1).

Figure 1
Il n’y a aucun compartiment et en particulier pas de noyau. C’est ce que l’on appelle un procaryote. Toutes les autres cellules, de la levure de boulanger à nos propres cellules sont des eucaryotes, elles possèdent un noyau [1]. C’est très important, car cela peut permettre de trouver des molécules spécifiques contre les bactéries sans trop de dommages pour les cellules de l’hôte, dont l'ADN est à l'abri dans le noyau.
Ce « sac » est une enveloppe constituée d'une membrane interne, composée essentiellement de lipides, mais dans laquelle peuvent s'insérer des protéines ou d'autres éléments. Elle est protégée par une paroi dont le principal élément est le peptidoglycane. C'est un polymère formé de longues chaînes de sucres pontées par de courtes chaînes peptidiques. On admet qu'il contribue à la forme et la rigidité de la bactérie et protège la membrane interne des effets de la pression osmotique(i). On peut le schématiser comme un gros grillage relativement facile à franchir (fig. 2). La taille d'une bactérie est de l'ordre de 1µm.

Figure 2
Les bactéries sont apparues sur terre il y a 3 milliards d'années(ii). On connaît environ 5000 espèces bactériennes, dont quelques dizaines sont pathogènes. On distingue deux types de bactéries, dites à Gram positif (Gram +) ou à Gram négatif(iii) (Gram -). Les Gram + (fig. 2a) ont une épaisse couche de peptidoglycane. Chez les Gram - (fig. 2b) le peptidoglycane est plus mince, mais est entouré d'une membrane externe complexe et difficile à franchir, sauf par des canaux hydrophiles, les porines, servant à l'entrée des nutriments.
On a aussi les mycobactéries, responsables de la tuberculose, de la lèpre, particulièrement bien protégées par leur enveloppe complexe et riche en lipides (25% environ, contre 1 à 2% pour les autres bactéries) (fig. 2c). En outre, leurs porines sont particulièrement étroites.
L'infection
Les bactéries sont responsables d'infections quand elles pénètrent et se multiplient dans un endroit de l'organisme où elles ne devraient pas être. Cela correspond à une rupture de l’équilibre entre bactérie et moyens de défense de l’hôte. Une infection peut être localisée(iv), locorégionale(v), septicémique(vi). On peut aussi les classer en infections communautaires(vii), et nosocomiales(viii).
L'infection peut être plus ou moins grave selon l'hôte infecté : une banale coupure nettoyée par lavage à l'eau et au savon sera sans conséquence pour un adulte en bonne santé, mais il existe de plus en plus de personnes à risque par suite des progrès de la médecine : personnes de plus en plus âgées, immunodéprimées (ayant subi une greffe par exemple), porteuses de prothèses, souffrant de maladies chroniques... La gravité dépendra aussi de la nature et des propriétés du germe infectieux : faculté d'adhérer aux cellules de l'hôte(ix), excrétion de toxines, capacité de survie dans la cellule infectée...
La lutte contre ces bactéries sera une véritable guerre sans merci, puisque, malgré les nombreuses armes dont nous disposons (les antibactériens), l'ennemi (la bactérie) possède une faculté de dissémination considérable, certaines pouvant se diviser toutes les 20 minutes ! En outre, elle possède plusieurs stratagèmes pour résister à nos armes [2]. Les bactéries communiquent entre elles, et communiquent aussi avec les cellules eucaryotes. Elles sont ainsi capables de maîtriser leur environnement pour y survivre ou s'y multiplier.
Les antibactériens
Quand on parle de la lutte contre les bactéries, il vaut mieux utiliser le terme d'antibactériens, bien spécifique, plutôt que celui plus couramment utilisé d'antibiotiques qui désigne les substances capables de combattre les microbes en général (bactéries, virus, parasites, champignons...). Par définition, les agents antibactériens inhibent la croissance bactérienne (on dit qu'ils sont bactériostatiques), ou tuent les bactéries (bactéricides).
Les figures 3 et 4 montrent quelques antibactériens importants. On pense souvent que la pénicilline est le premier antibactérien connu. En fait, très tôt, certains composés étaient utilisés contre les infections : eau de Javel(x) comme désinfectant [3], mercure contre la syphilis ; dès 1897, le médecin militaire Ernest Duchesne a traité avec succès des porcs atteints de typhoïde porcine par la moisissure Penicillium glaucum(xi). En 1911, avec le chimiste Alfred Bertheim et le bactériologiste Sahachiro Hata, Paul Ehrlich met au point le Salvarsan, actif contre la syphilis. En 1936, à l'Institut Pasteur de Paris, Thérèse et Jacques Tréfouël [4] synthétisent les sulfamides (fig 2), actifs contre de nombreux germes. Quant à la pénicilline, découverte par sérendipité [5] en 1928 par Fleming, son intérêt thérapeutique n'apparut que vers 1939(xii).

Figure 3. Antibactériens découverts par sérendipité

Figure 4. Exemples d’antibactériens
Le tableau 1 montre les dates d'apparition des principaux antibactériens, et leur origine. La plupart sont d'origine naturelle, mais des dérivés sont ensuite préparés par synthèse totale ou par hémisynthèse(xiii).
Pour combattre une infection, un antibactérien doit :
- entrer dans la bactérie et ne pas en être expulsé ;
- échapper à des mécanismes d'inactivation (enzymes) ;
- atteindre sa cible et la perturber en causant un dommage à la bactérie.
Cibles des antibactériens
Selon l'antibactérien, ce sont différents processus indispensables à la vie d'une bactérie que les antibactériens peuvent inhiber :
- Inhibition de la synthèse du peptidoglycane : la bactérie ne pourra plus fabriquer son enveloppe
- β lactames (pénicilline) et glycopeptides (vancomycine) ;
- Inhibition de la synthèse des acides nucléiques : la bactérie ne peut plus se multiplier
- ARN : rifampicine,
- ADN : quinolones (ofloxacine).
- Inhibition de la synthèse des protéines : la bactérie ne fabrique plus de protéines ou en produit de défectueuses. Elle n'aura plus les enzymes nécessaires à son métabolisme, par exemple :
- aminoglycosides (streptomycine),
- macrolides (érythromycine),
- tétracyclines,
- chloramphénicol.
En conclusion, même si nous disposons comme on le voit, d'un important arsenal chimique [6] pour lutter contre les maladies et en particulier les infections, la bataille est cependant difficile, des maladies comme la tuberculose, que l'on pensait quasiment éradiquée, continuent à exister, même dans les pays développés. Et il faut en plus compter avec la résistance que les bactéries peuvent développer. Sur ce sujet, consultez le Zoom sur la résistance des bactéries aux antibactériens.
Tableau 1 : Apparition de quelques antibactériens
| Classe | Date de la découverte → date d'élucidation de la structure | Origine |
| β-lactames: pénicillines | 1929 → 1945 | F |
| Sulfonamides | 1936 | S |
| Aminoglycosides | 1944 → 1947 | F |
| Phénicolés (chloramphénicol | 1948 → 1949 | F S |
| Peptides | 1948 → 1964 | F |
| β-lactames: céphalosporines | 1948 → 1961 | F |
| Macrolides | 1952 → 1965 | F |
| Tétracyclines | 1953 → 1962 | F |
| Streptogramines | 1955 → 1966 | F |
| Nitro imidazoles | 1960 | S |
| Rifampicine | 1960 → 1973 | F |
| Lincosamides | 1962 → 1964 | F |
| Acide fusidique | 1962 → 1965 | F |
| Quinolones | 1962 | S |
| Triméthoprime | 1962 | S |
| Glycopeptides | 1962 → 1983 | F |
| Fosfomycine | 1969 → 1969 | F S |
| Inhibiteurs de β-lactamases | 1975 → 1976 | F S |
| β-lactames: carbapénèmes | 1976 → 1978 | F S |
| Monobactames | 1979 → 1981 | F S |
F: obtenu par fermentation (produit naturel)
S: obtenu par synthèse
.jpg)
E. coli
(i) Pression qui détermine le phénomène d'osmose et qui correspond à la différence de pressions exercées de part et d'autre d'une membrane semi-perméable par deux liquides de concentration différente.
(ii) Les premiers hommes il y a seulement 10 millions d'années.
(iii) Hans Gram est un bactériologiste danois de la seconde moitié du XIXe siècle, qui a mis au point une coloration permettant de distinguer grâce à leur membrane deux types de bactéries : celles à Gram positif (Gram +) se colorent en rose, celles à Gram négatif (Gram -) ne se colorent pas.
(iv) Par exemple sur un doigt.
(v) Tout le membre.
(vi) Diffusée à tout l'organisme à partir du foyer initial : très grave.
(vii) Survenant à l'extérieur d'un établissement de santé.
(viii) Qui n'existaient pas chez le malade à son entrée à l'hôpital.
(ix) Lors d'une infection urinaire, on recommande de boire beaucoup : ainsi, si les germes infectieux sont peu adhérents, ils seront facilement éliminés.
(x) Hypochlorite de sodium, dont la découverte est attribuée à Claude Louis Berthollet en 1775, dans son usine du quartier de Javel à Paris.
(xi) Il a publié ce résultat sans savoir quelle substance était produite par son Penicillium.
(xii) On reverra avec intérêt le film « Le troisième homme ».
(xii) En partant de la molécule naturelle ou d'un précurseur.
Pour en savoir plus
[1] Parasite, champignon, bactérie et virus : quelles différences ?, N. J. Moreau, Question du mois, Mediachimie.org (2020)
[2] On regardera avec profit et plaisir la vidéo Bactéries, nos amies ? CERIMES (1 janvier 1990) Canal-u.tv
[3] Pourquoi ne pas mélanger de l’eau de Javel et du détartrant ?, F. Brénon,Question du mois, Mediachimie.org (2022)
[4] Thérèse et Jacques Tréfouël, le binôme indissociable, Institut Pasteur - Notre histoire
[5a] Il était une fois la sérendipité, S. Allemand et S. Catellin, Le Media Paris-Saclay (2014)
[5b] La sérendipité, un chemin de traverse à suivre, C. Monneret L'Actualité Chimique n°385 (mai 2014)
[6] Chimie et médicaments : un bel avenir !, B. Meunier, Colloque chimie et nouvelles thérapies (novembre 2019)
Crédits :
- illustration E. coli : Image par Gerd Altmann / Pixabay
- figures : © NJ Moreau
Actuellement en France, un homme sur deux et une femme sur trois seront atteints d’un cancer dans leur vie… on dénombre environ 380.000 nouveaux cas de cancer par an et on enregistre 140.000 décès ! [1].
Le cancer est une maladie complexe qui résulte d’une prolifération anormale des cellules. Comme chacun sait, il n’y a pas un cancer mais des cancers et ceci impose donc de posséder des stratégies curatives adaptées à chaque type de cancer. Les principales stratégies utilisées dans le traitement des cancers sont la chirurgie, la radiothérapie, la chimiothérapie, les thérapies ciblées, et l’immunothérapie.
La chimiothérapie est la méthode la plus ancienne utilisée pour essayer de détruire les cellules tumorales par administration de médicaments dits « cytotoxiques ». Ces médicaments peuvent agir sur différents processus impliqués dans la multiplication des cellules.
D’abord certains médicaments empêchent la réplication de l’ADN et sa transcription en ARN et peuvent être par exemple : 1) des agents alkylants (chlorméthine) 2) des intermédiaires électrophiles (dérivés du platine) 3) des intercalants entre les bases nucléiques dans la double hélice de l’ADN (daunorubicine). On utilise aussi des antimétabolites (5-fluoro-uracile) qui s’incorporent dans l’ADN à la place des bases nucléiques de l’ADN. On emploie parfois des inhibiteurs enzymatiques empêchant des réactions conduisant au développement des cellules (méthotréxate).
Enfin d’autres molécules altèrent la mitose (division cellulaire) ; ce sont des alcaloïdes comme la vinblastine, la navelbine ou des hétérocycles comme le taxotère. Ces dernières molécules se sont révélées très efficaces contre les cancers du sein mais aussi du poumon et ont été synthétisées par l’équipe de Pierre Potier dès les années 80 à l’ICSN de Gif-sur-Yvette dans la région parisienne [2].
Un protocole de chimiothérapie fait souvent appel à une association de plusieurs médicaments qui agissent sur ces différents processus. Les chimiothérapies sont souvent redoutées en raison de leurs effets secondaires (chute des cheveux, nausées, vomissements, baisse du nombre de cellules sanguines…). En effet, ces molécules s’attaquent aux cellules en développement rapide telles que les cellules tumorales mais aussi aux cellules saines qui se multiplient activement comme celles des cheveux, du sang ou des muqueuses digestives. Plus récemment des approches plus spécifiques des cellules tumorales ont été développées.
Les thérapies ciblées constituent une autre famille de traitements du cancer le plus souvent disponibles par voie orale. En ciblant spécifiquement certaines molécules de l’organisme, elles bloquent des mécanismes qui sont indispensables à la prolifération des cellules cancéreuses ou, plus globalement, au développement de la tumeur. Certains agissent sur les cellules cancéreuses à proprement parler et d’autres sur les cellules du micro-environnement tumoral, par exemple en bloquant la formation des vaisseaux sanguins qui irriguent une tumeur solide (médicaments antiangiogéniques) ou en activant des cellules immunitaires (immunothérapie). Selon les cancers, les thérapies ciblées peuvent être prescrites seules, en association entre elles ou avec d’autres traitements [3]. En éliminant avant tout les cellules porteuses de l’anomalie moléculaire ciblée, ces thérapies sont généralement mieux tolérées que les chimiothérapies conventionnelles mais elles ne sont pas exemptes d’effets indésirables.
L’immunothérapie regroupe un ensemble de stratégies visant à mobiliser ou à renforcer les défenses immunitaires des patients de manière à ce qu’elles s’attaquent aux cellules tumorales.
L’immunothérapie « spécifique » consiste à bloquer spécifiquement des protéines à la surface des cellules cancéreuses ou dans leur micro-environnement pour freiner la croissance tumorale. Elle repose notamment sur l’utilisation d’anticorps monoclonaux (les anticorps sont des protéines produites par le système immunitaire pour neutraliser et signaliser des éléments anormaux présents dans l’organisme). Un anticorps monoclonal est spécifique d’une seule cible, il se lie fortement à l’antigène (un antigène est une molécule toxique (virus, bactérie…), présente dans l’organisme (sang) qui stimule une réponse immunitaire). Les anticorps monoclonaux sont des anticorps qui n’existent pas naturellement dans l’organisme. Ils sont produits en laboratoire à partir de cellules-mères (animales) sélectionnées en culture à partir de levures ou de bactéries pour bloquer des mécanismes spécifiques, essentiels aux cellules cancéreuses. Cette méthode a fait l’objet de l’attribution du Prix Nobel de Médecine en 1984 au britannique César Milstein, au danois Niels Jerne et au suisse Georges Köhler. De nombreux anticorps monoclonaux sont sur le marché ; ils reconnaissent des protéines de surface spécifiques de la cellule tumorale et bloquent leur activité conduisant à la mort de la cellule et la reconnaissance de cette cellule par le système immunitaire. Historiquement, les premiers exemples de cette classe de traitements sont le bevacizumab, un « antiangiogénique » (il bloque le signal qui stimule la production de nouveaux vaisseaux sanguins dans les tumeurs) et le trastuzumab, un « anti HER2 » utilisé avec succès notamment pour traiter les cancers du sein dits HER2 positifs [4].
Une autre stratégie consiste à réactiver les lymphocytes T (globules blancs fabriqués dans le thymus (T), glande située à la base du cou) du système immunitaire qui sont désactivées par certaines cellules tumorales.


L’administration d’un anticorps monoclonal dirigé contre les protéines de la cellule tumorale qui inhibent l’activité des cellules T permet de retrouver un système immunitaire actif qui détruit la cellule tumorale.
Les immunoconjugués
Le concept est de « marier » la spécificité des anticorps monoclonaux à la puissance d’un agent anticancéreux pour augmenter l’efficacité de l’un et l’autre et diminuer les effets secondaires si possible. Ces immunoconjugués (en français) ou antibody-drugs conjugates (ADC) (en anglais) sont des « prodrogues » constituées par une molécule toxique (cytotoxique, toxine ou élément radioactif), et un anticorps qui sont reliés par un lien chimique (« linker » en anglais) réalisé par des liaisons chimiques covalentes.
Le mécanisme d’action de ces immunoconjugués est : 1) la fixation de l’anticorps sur la surface de la cellule cancéreuse grâce à une interaction anticorps-antigène, 2) l’endocytose (pénétration) dans le cytoplasme de la cellule conduisant à une structure appelée endosome accompagnée d’une acidification du milieu, 3) une coupure du lien par des enzymes permettant de libérer la molécule cytotoxique dans la cellule cancéreuse.
Signalons que les molécules cytotoxiques utilisées doivent être 100 à 1000 fois plus puissantes que les molécules classiques de la chimiothérapie car le greffage des molécules cytotoxiques sur l’anticorps est limité pour des raisons stériques à un certain nombre.
L’agent de liaison joue un rôle primordial, il doit : 1) être suffisamment stable pour éviter la libération de l’agent cytotoxique dans le sang ce qui conduirait à des effets toxiques indésirables, 2) permettre la libération de l’agent toxique uniquement dans la cellule cancéreuse. La réunion de la molécule antitumorale avec l’anticorps s’effectue par un lien chimique fonctionnalisé. Une fonction de ce lien permet une réaction avec certaines fonctions polaires de l’agent antitumoral et, du côté anticorps, une autre fonction du lien va réagir avec certains acides aminés de l’anticorps tels que des lysines ou cystéines. Les liaisons sont de type peptidique donc hydrolysables par des enzymes ou de type ponts disulfures qui peuvent alors être réduits[5]. L’immunoconjugué, grâce à la reconnaissance spécifique d’une protéine de surface de la cellule tumorale amène l’agent toxique majoritairement sur les cellules tumorales ciblées évitant ainsi les effets sur les cellules saines [6].
Une autre technique pour éviter les effets indésirables est que les immunoconjugués peuvent être encapsulés dans des microgouttelettes (utilisation de la microfluidique pour produire les microgouttelettes), qui sont injectées par voie intraveineuse et acheminées vers la tumeur grâce au flux sanguin. Le largage de l’immunoconjugué se fait au niveau de la tumeur grâce à des ultra-sons à l’endroit voulu et au moment voulu, la tumeur étant repérée grâce à un échographe. C’est ce qui a été réalisé, en particulier, avec la prodrogue [monométhylauristatine]. Les immunoconjugués actuellement en essai clinique ainsi que la mise au point de nouvelles techniques [7] permettent d’envisager des activités antitumorales sur les cancers du sein de grade élevé et des leucémies aigües avec une bonne efficacité et en minimisant les effets secondaires.
NB : L’auteur tient à remercier vivement Nicole Moreau, Janine Cossy et Jean-Marc Paris pour leurs aides à la rédaction de ce texte et leurs encouragements.
Références bibliographiques
[1] Biologie de synthèse : une nouvelle voie pour le traitement du cancer, C de Obaldia, colloque Chimie et Biologie de synthèse (14 février 2018), conférence et article (Mediachimie.org)
[2] Recherche et découverte de nouveaux médicaments antitumoraux : la Navelbine et le Taxotère, P. Potier, L'Actualité Chimique (janvier-février 1995) n° 185 p. 5
[3] Les anticorps monoclonaux : un fantastique arsenal thérapeutique en plein devenir, M. Fougereau, medecine/sciences (2009) 25:997–998
[4] Petites et grosses molécules innovantes dans le traitement des cancers, J.-P. Armand, colloque Chimie et nouvelles thérapies (13 novembre 2019), conférence et article (Mediachimie.org)
[5] Un point sur : Les immunoconjugués en oncologie (fiche 45) L. Gauzy-Iazo, L'Actualité Chimique (décembre 2016) n° 413 p. 63
[6] La montée en puissance des immunoconjugués en oncologie, Une liaison réussie entre un anticorps et une petite molécule cytotoxique, E. Vigne et I. Sassoon, medecine/sciences (2014) 20:855-863
[7] Nature et chimie : des alliées pour accéder à de nouveaux médicaments, J. Cossy, colloque Chimie et nouvelles thérapies (13 novembre 2019), conférence et article (Mediachimie.org)
Crédit illustration : Catharanthus roseus, Vengolis, CC BY-SA 4.0, WikiMedia
L’électrolyse de la vapeur d’eu est réalisée autour de 800°C. La réaction est réversible car la cellule peut fonctionner comme électrolyseur mais aussi sous forme de pile à combustible. La technologie devient progressivement mature ; ainsi la startup SYLFEN crée en 2015 par le CEA utilise ce type d’électrolyse pour le chauffage d’immeubles autonomes en énergie.
Source : L’Actualité Chimique n° 466 (octobre 2021) p. 12-19
L’électrolyse à membrane échangeuse de protons utilise une membrane à base de polymère d’acides perfluorosulfoniques (PFSA). La membrane a un double rôle : elle permet de séparer les gaz hydrogène et oxygène formés et assure le transfert des protons de l’anode vers la cathode. Le principe de fonctionnement y est exposé en détail, notamment les procédés électro-catalysés au niveau des électrodes. Le gaz dihydrogène produit est de grande pureté. Le procédé est en plein développement pour devenir rentable à moyen terme.
Source : L’Actualité Chimique n° 466 (octobre 2021) p. 20-27
On dénombre 14 millions d’accidents vasculaires cérébraux (AVC) par an dans le monde ! L’implantation de matériaux biocompatibles pour libérer des molécules pharmacologiques directement dans le cerveau permet alors de reconstruire des tissus. Or les hydrogels sont des réseaux 3D de chaînes polymériques réticulés gonflés en eau jusqu’à 80-90 % en eau. Ils sont injectables car liquides à la température corporelle ; de plus ils sont dégradables, non toxiques et peuvent être chargés en molécules thérapeutiques. Le polymère utilisé est un copolymère bloc entre le PLA (acide polylactique pour le caractère hydrosoluble), le PNIPAM (poly(N-isopropylacrylamide) pour le caractère injectable) et le PEG (polyéthylène glycol pour la prise en eau) ; il y a autoassemblage à température ordinaire avec un cœur hydrophobe de PLA et une couronne hydrophile de PNIPAM et de PEG. Ce gel peut solubiliser à la fois des produits hydrophiles et hydrophobes : ainsi le riluzole, introduit dans le gel qui, injecté dans le cerveau, inhibe la libération de glutamate toxique pour les neurones. Les essais cliniques sont encore au tout début du processus.
Source : L’Actualité chimique n° 451 (mai 2020) pp. 63-64
Voilà 100 ans, le 11 janvier 1922, que pour la première fois un enfant de 14 ans en coma diabétique reçut une injection d’un extrait de pancréas (et donc d’insuline) ; il fut sauvé et survécu 13 ans avec des injections régulières d’insuline.
Quel est le rôle de l’insuline ?
L’insuline est une hormone (i), secrétée par le pancréas, dès que la glycémie (taux de glucose dans le sang) dépasse un seuil. Cette sécrétion favorise un retour de la glycémie à une valeur de base. Son absence ou sa sécrétion insuffisante est responsable des divers types de diabète.
Quelle est la structure de l’insuline ?
L’insuline est une protéine. Sa structure a été décrite en 1955 par Frederick Sanger (ii) et représentée en 3 D par Dorothy Hodgkin (iii) en 1969.
De formule brute C257H383N65O77S6, sa masse molaire vaut 5807 g.mol-1. La structure de l’insuline est complexe. Un premier brin appelé chaine A contient 21 acides aminés (acides aminés notés aa par la suite). Un deuxième brin appelé chaine B contient 30 aa. Ces 2 chaines sont reliées entre elles par 2 ponts disulfures (S-S) entre 2 cystéines de A et 2 cystéines de B. Il existe aussi un pont S-S interne entre deux cystéines de la chaine A. L’ensemble des 2 chaines forme un monomère. Voir image ci-dessous.

Trois dimères se forment grâce à des liaisons hydrogènes entre des aa des chaines terminales de B et sont assemblés entre eux via des interactions avec deux ions Zn(II). L’insuline a donc une structure hexamère.
Insuline humaine et animale
La structure de l’insuline est particulière à l’espèce, mais celles de l’homme, du porc et du bœuf sont très voisines.
L’insuline humaine est différente de l’insuline porcine juste par un aa de la chaine B en position 30. Celle du bœuf diffère en plus par 2 aa de la chaine A en positions 8 et 10.
Les premières insulines administrées aux diabétiques insulino-dépendants
Elles sont extraites de pancréas de bœuf ou de porc, mise en solution acide (pH = 3) et malheureusement imparfaitement purifiées. Le patient doit avoir 3 ou 4 injections par jour. Les impuretés et la différence structurelle de ces insulines sont responsables de réactions locales ou d’allergies. Ainsi, de 1922 jusqu’en 1999 (en France) les diabétiques ont pu recevoir des insulines extraites du porc et du bœuf. Depuis les années 80, les insulines reçues par les diabétiques ont beaucoup évolué.
L’insuline porcine modifiée appelée insuline semi-synthétique humaine
Au début des années 80, l’insuline porcine a subi des transformations enzymatiques permettant de remplacer l’aa alanine en position 30 de la chaine B par l’aa thréonine ; ainsi on obtient une insuline semi-synthétique humaine. Bien qu’apportant au malade une plus grande efficacité, ce procédé nécessitait encore un approvisionnement en insuline porcine et provoquait dans certains cas des réactions de rejet.
Des insulines différenciées pour améliorer rapidité et durée d’action
La notion d’insuline différenciée est liée à la vitesse et la durée d’action. On parle d’insulines, rapides, intermédiaires ou lentes.
Ainsi des insulines rapides sous forme d’hexamères sont injectées pour éviter une augmentation de la glycémie trop importante au moment des repas, tandis que des insulines intermédiaires permettant de combler les besoins humains en insuline indépendamment des repas, sont injectées 2 à 3 fois par jour.
Ces insulines intermédiaires, nommées NPH (Neutral Protamine Hagerdorn), proposées dès 1923 par Hagerdorn, contiennent en plus de l’insuline humaine, des protamines (iv), une quantité variable d’ions Zn2+ et un milieu neutre grâce à un tampon phosphate. Cette formulation injectée diffuse alors lentement dans le corps humain.
L’inconvénient de ces premières insulines différenciées était leur délai d’action et la durée.
L’insuline humaine obtenue par le génie génétique appelée insuline biogénétique humaine ou recombinante
Dès le début des années 80 des insulines humaines obtenues par génie génétique sont apparues sur le marché et depuis 1999 (en France) seules ces insulines sont utilisées.
Très schématiquement, un gène (portion d’ADN) codant pour l’insuline humaine est inséré dans l’ADN d’une cellule hôte, la bactérie Escherichia coli. Celle-ci produit alors de l'insuline humaine grâce au gène supplémentaire intégré. Pour la production de grandes quantités, cette bactérie modifiée est introduite dans un fermenteur où elle se multiplie et produit le composé recherché. Des étapes de séparation et purification sont indispensables.
Pour concrétiser on peut citer pour exemples, deux modes de fabrication industrielle d’insuline recombinante. Ce ne sont pas les seuls.
Dans le premier, il est d’abord créé indépendamment, par génie génétique, les 2 chaines A et B rallongées volontairement par quelques aa dont la présence facilite l’étape chimique ultérieure. À l’issue de ces fermentations ces chaines sont séparées par ultrafiltration ou centrifugation. La création des ponts sulfure (S-S) entre les 2 chaines est alors réalisée par voie chimique. Une chromatographie préparative à basse pression par échanges d’ions est alors réalisée. Une nouvelle étape de réaction chimique ou enzymatique permettant la découpe des aa excédentaires est suivie d’une ultrafiltration puis d’une chromatographie préparative à haute pression (70 bars) sur gel de silice greffé ou non, pour conduire à l’insuline humaine recombinante purifiée à usage pharmaceutique.
Dans le deuxième mode, la bactérie est programmée pour reproduire au cours de la fermentation les chaines AB accrochées entre elles en une seule chaine. Après une séparation / filtration, une découpe par voie chimique ou enzymatique et la création des ponts sulfure sont réalisées suivies d’une chromatographie par échange d’ions. Une purification finale par chromatographie HP est également nécessaire.
Le génie génétique permet aussi de produire des insulines aux actions différenciées.
Les insulines analogues rapides ou lentes
La fin des années 90 a vu arriver les insulines dites analogues rapides et lentes. Elles sont aussi issues du génie génétique. Elles ont pour objectif de modifier la cinétique ou la solubilité de l’insuline.
Ce sont des insulines très légèrement différentes de l’insuline humaine par changement de quelques aa (addition ou soustraction ou échange) et de quelques adjonctions.
On distingue les insulines analogues rapides, à début d’action plus rapide et durée d’action plus courte que l’insuline humaine (lispro (v), en 1996, asparte (vi) en 2000 ou glulisine (vii) en 2004) qui par juste quelques changements (un aa par un autre) permettent de garder la même action au niveau de la glycémie. La rapidité d’action est due au fait que ce n’est plus l’hexamère qui est formé mais le monomère.
Inversement les insulines analogues lentes ou à action prolongée (glargine (viii) en 2000 et détémir (ix) en 2004) par des modifications d’aa permettent un changement du point isoélectrique. Solubles en milieu acide ces insulines précipitent au pH physiologique et les hexamères formés ont ainsi une durée d’action beaucoup plus longue.
On peut citer quelques fabricants d’insuline dans le monde : Novo Nordisk (danois) leader mondial, Eli Lilly (américain), Sanofi-Aventis (français), Biocon en Inde, Wanbang Biopharma en Chine, Julphar au Moyen Orient…
Proche de nous, on trouve des sites de production d’insuline à Chartres (Novo Nordisk), à Fegersheim (Lilly) et Francfort (Sanofi).
D’après le rapport 2019 de la FID (Fédération Internationale du Diabète), « 578 millions d'adultes seront atteints de diabète d'ici 2030 et 700 millions d'ici 2045 ». On mesure l’importance du savoir-faire indispensable pour la production d’insuline.
Aujourd’hui les axes de recherche dans l’amélioration du traitement sont tournés non plus vers une production d’insuline toujours plus active, mais plutôt dans des injections plus efficaces : pompes en boucle fermée avec mesure en continu de la glycémie et injection en continu d’insuline via l’intervention de l’Intelligence Artificielle (c’est ce qu’on nomme le pancréas artificiel). D’autres nouvelles thérapies sont en cours d’étude : ainsi des cellules souches (implantées directement sous la peau) se transforment en cellules béta des îlots de Langerhans (les cellules pancréatiques productrices d’insuline) et produisent de l’insuline directement utilisable par le corps humain.
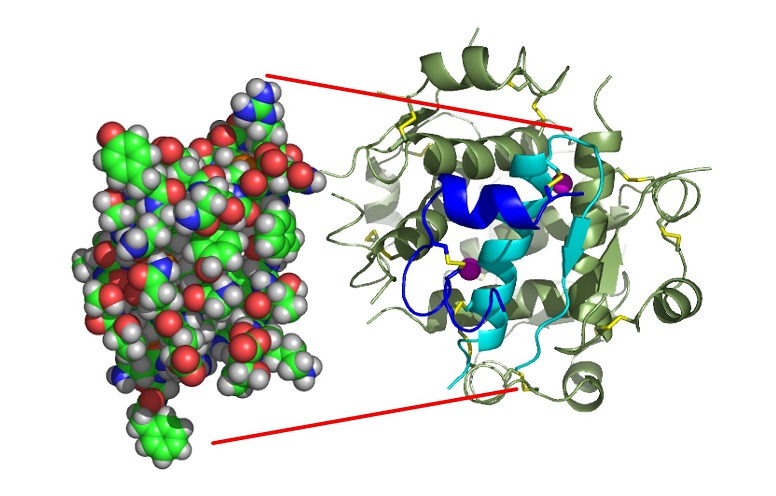
Monomère insuline humaine
(i) En 1922 deux biologistes canadiens Frederick Banting et Charles Best découvrent l’insuline par extraction du pancréas.
(ii) Frederick Sanger a eu le prix Nobel de Chimie en 1958 pour son travail sur la structure des protéines, particulièrement celle de l’insuline.
(iii) Dorothy Hodgkin a eu le prix Nobel de Chimie en 1964 pour « sa détermination par des techniques aux rayons X des structures de substances biochimiques importantes », ce qui lui permit de déterminer la structure tridimensionnelle de l’insuline en 1969.
(iv) Les protamines sont des petites protéines nucléaires (trouvées au centre de noyaux cellulaires) riches en aa arginine.
(v) Insuline lispro : la molécule comporte une inversion de deux aa en bout de chaîne B, qui ne modifie pas la liaison au récepteur, mais bloque la formation de dimères et d'hexamères d'insuline.
(vi) Insuline asparte : c'est une insuline analogue où un seul acide aminé a été modifié, en particulier une proline avec un acide aspartique à la position B28.
(vii) Insuline glulisine : insuline analogue où l’aa asparagine en position B3 est remplacé par la lysine et la lysine en position B29 est remplacée par l'acide glutamique.
(viii) Insuline glargine : remplacement de l'asparagine par la glycine en position 21 de la chaîne A et par l'extension carboxy-terminale de la chaîne B par 2 résidus arginine.
(ix) Insuline détémir : délétion de la thréonine en position B30 et fixation sur la lysine en position B29 de l’acide tétradécanoïque (ou myristique) de formule CH3–(CH2)12–COOH, ce qui lui permet de se complexer à l'albumine dans le sang. Puis lentement le complexe se dissocie et libère l’insuline.
Pour en savoir plus
(1) Histoire de l’insuline : entre le biologique et l’artificiel, G. El Mghari, S.Baki et N. El Ansari, Service d’endocrinologie, Laboratoire PCIM, Université Cadi Ayyad, Marrakech, Hegel vol. 4 n°2 (2014) p. 208 (DOI : 10.4267/2042/53793)
(2) L’insuline produit du jour Société chimique de France
(3) Les 90 ans de la découverte de l'insuline, par la Fédération Française des Diabétiques
(4) Les insulines, médicaments actuels et évolution dans la prise en charge du diabète insulinodépendant, Thèse Alexandre Bitil (2015), tout particulièrement les modes d’obtention pages 21 à 26 (HAL Id : dumas-01171688)
(5) Le génie génétique à la rescousse des diabétiques Musée Armand Frappier Canada
(6) État des lieux passé et actuel de l’insuline (thérapies, procédés) et perspectives d’évolution, Thèse d'exercice en pharmacie (Toulouse) de Delpech Romain (2015)
Crédits :
Illlustration : Monomère insuline humaine par Isaac Yonemoto. Transféré de en.wikipedia.org vers Commons. Premier téléchargement vers en.wp par Takometer, CC BY 2.5, Lien
Figure : Insuline humaine : enchainement des acides aminés et ponts disulfure réalisée par Lydie Amann. DR.